 ¿Qué es la alfa-1-antitripsina?
¿Qué es la alfa-1-antitripsina?
El Alfa-1 Antitripsina (AAT) es una proteína que se produce en el hígado que tiene como función principal proteger los pulmones del deterioro e inflamación causadas por agentes externos (tabaco, otras sustancias inhaladas…) que son contaminantes e irritantes y/o por infecciones que atacan el tejido pulmonar.
En condiciones normales esta proteína pasa de las células hepáticas al sistema la sangre y es repartida por todo el organismo. Cuando este traspaso no se lleva a cabo de la manera habitual la proteína se acumula en el hígado pudiendo provocar enfermedades hepáticas; y un descenso del AAT en sangre, que desprotege a los pulmones. Esta desprotección, unido a otros agentes como infecciones y tabaco, puede llegar a provocar enfermedades pulmonares. Entre estas patologías cabe destacar las siguientes:
- Cirrosis, hepatitis y colestasis neonatal en el hígado.
- Bronquiectasia, enfisema, bronquitis crónica y asma no reversible en el pulmón.
- Vasculitis tipo C-Anca positivo y paniculitis, enfermedad de la piel menos frecuente.
- Fibromialgia, en estudio.
Prevalencia
Se estima que 1 de cada 2.500 personas está afectada por el Déficit de Alfa 1 Antitripsina. En España hay alrededor de 22 casos por cada 100.000 habitantes.
Es una enfermedad infradiagnosticada que está incluida dentro del grupo de Enfermedades Raras
¿Cuáles son las causas de este déficit?
Las variantes genéticas que implican algún tipo de alteración se denominan con las letras S y Z, respectivamente. Una persona que tenga un gen normal o M y otro deficitario, ya sea Z o S, serán portadores de esta enfermedady podrán transmitir su gen deficitario a sus descendientes. Estas personas presentarán un descenso de actividad de la proteína que puede tener consecuencias leves en ellos.
En los casos en que ambos alelos son deficientes, es decir, SS, ZS o ZZ, la enfermedad se desarrollará con diferentes grados de gravedad, en función de la combinación, siendo la ZZ la combinación con mayores consecuencias, como puede observarse en el siguiente cuadro:
| Déficits más frecuentes | Cantidad de AAT | Riesgo de Enfermedad hepática | Riesgo de enfisema |
| MS | Disminución muy leve | No | No |
| SS | Disminución leve | No | No |
| MZ | Disminución leve/moderada | Ligero | Muy ligero aumento |
| ZS | Disminución moderada | Moderado | Moderado |
| ZZ | Disminución grave | Alto | Alto |
Diagnóstico
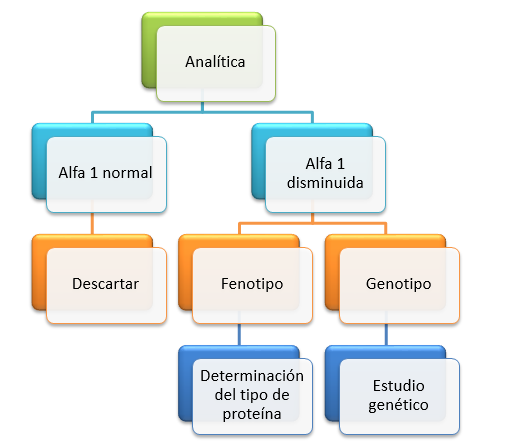
¿Quién debe hacerse las pruebas?
El déficit AAT es la causa hereditaria más común para la EPOC y la enfermedad hepática infantil.
Todavía hay mucho infra diagnóstico de esta condición en el mundo. Según la OMS, (Organización Mundial de Salud, la ATS (American Thoracic Society) y la ERS (European Respiratory Society) se recomienda la prueba a todas las personas con las siguientes patologías:
Respiratorias:
- EPOC
- Enfisema
- Bronquiectasia
- Bronquitis crónica
- Asma no reversible en el post-tratamiento
Hepáticas:
- Enfermedad hepática de origen desconocida
- Enfermedad hepática crónica
- Cáncer del hígado (adulto)
También es aconsejable en:
- Pacientes con paniculitis o vasculitis de causa desconocida
- Familiares consaguíneos de una paciente diagnosticado de déficit de AAT
- Pacientes que se hayan realizado un proteinograma que muestre disminución de alfa globulinas
- A todas las personas cuyos familiares han padecido alguna enfermedad de este tipo de origen desconocido.
Es importante tener en cuenta que no todas las personas que tienen la condición genética del déficit AAT padecen una enfermedad relacionada con ella. Existen otros factores probablemente de origen genético o ambiental que contribuyen a su desarrollo. Todavía se desconocen otras posibles causas.
Tratamiento
No hay tratamiento del déficit genético por lo que no tiene cura, pero sí se deben de tratar las diferentes manifestaciones que presenta para mejorar la evolución y la calidad de vida de estos pacientes. Algunas recomendaciones son:
- Cambios estilo de vida.
- No fumar.
- Evitar exposición a agentes dañino.
- Ejercicio:
- Adaptado a la capacidad física del sujeto y siguiendo los consejos médicos.
- Dieta sana:
- Evitarse el sobrepeso y la desnutrición.
- Afecciones hepáticas:
- Tratamiento farmacológico.
- Trasplante de hígado: en casos muy graves.
- Afecciones pulmonares:
- Disnea:
- Inhaladores u otros medicamentos.
- Enfisema:
- Oxigenoterapia.
- Trasplante pulmonar.
- Administración de Alfa 1 antitripsina:
- Frena el deterioro pulmonar.
- No tiene efectos a nivel hepático.
- Disnea:
Más información
Asociación de pacientes Alfa-1 de España www.alfa1.org.es
SEPAR. Guía para familiares de niños con DAAT. www.separ.es
Registro Nacional de Enfermedades Raras del Instituto de Salud Carlos III https://registroraras.isciii.es Contiene información general sobre diversas enfermedades raras y un apartado para que los afectados se registren.
Asociaciones
Asociación Alfa-1 de España.
R/ Xosé Chao Rego, 8 Baixo
15705 Santiago de Compostela
Teléfono: 981 555 920
Contacto: info@alfa1.org.es
Web: www.alfa1.org.es
Asociación Alfa-1 de Europa
Teléfono: 965 966 553
Contacto: chair@apha1awareeness.org.uk
Web:www.alphaeurope.eu/component/option,com_frontpage/Itemid,1/lang,en/